library(NMF)
## Loading required package: registry
## Loading required package: rngtools
## Loading required package: cluster
## NMF - BioConductor layer [OK] | Shared memory capabilities [NO: bigmemory] | Cores 2/2
## To enable shared memory capabilities, try: install.extras('
## NMF
## ')
##
## Attaching package: 'NMF'
## The following object is masked from 'package:generics':
##
## fit
library(vegan)
## Loading required package: permute
## Loading required package: latticeKanazawa.R #3 での藤原先生のライトニングトークにあった非負値行列分解(Non-negative Matrix Factorization: NMF, 非負値行列因子分解とも)で生物群集の解析ができそうな気がしたので、ためしてみました。
準備
NMFパッケージをインストールします。が、BioconductorにあるBiobaseが必要といわれるので、まずそちらをインストールしておきます。
インストールできたら、読み込んでおきます。群集生態学の解析パッケージのveganも読み込みます。
データ
データには、“Numerical Ecology with R, 2nd ed.”(日本語訳: Rによる数値生態学)のDoubs川の魚のデータを用います。このデータは同書のサポートページからダウンロードできます。
load("data/Doubs.RData")読み込んだデータのうちのspeというデータフレームを使います。このデータフレームは、行がサンプリングサイト、列が魚種となっていて、値は個体数のランクで0〜5の値をとります。
データフレームの左上の部分を見てみます。
spe[1:10, 1:5]
## Cogo Satr Phph Babl Thth
## 1 0 3 0 0 0
## 2 0 5 4 3 0
## 3 0 5 5 5 0
## 4 0 4 5 5 0
## 5 0 2 3 2 0
## 6 0 3 4 5 0
## 7 0 5 4 5 0
## 8 0 0 0 0 0
## 9 0 0 1 3 0
## 10 0 1 4 4 0実行
標準化
データを標準化します。生物群集データでよく用いられる方法のひとつである、各行について2乗和が1になる方法で変換しています(decostand関数でmethod = "normalize")。
また、8番目のサイトはどの種も記録がないため削っておきます。
spe_norm <- decostand(spe[-8, ], method = "normalize")このデータフレームを行列Xとして
X ≒ RP
と分解します。
NMFの実行はnmf関数でおこないます。rank(潜在パターンの数)はここでは3としました。seed引数ではNonnegative Double Singular Value Decompositionをseeding methodとして使用することを指定しています。
spe_nmf <- nmf(spe_norm, rank = 3, seed = "nndsvd")結果
各サンプリングサイトの潜在パターンへの分解(R)をヒートマップでしめします。
basismap(spe_nmf)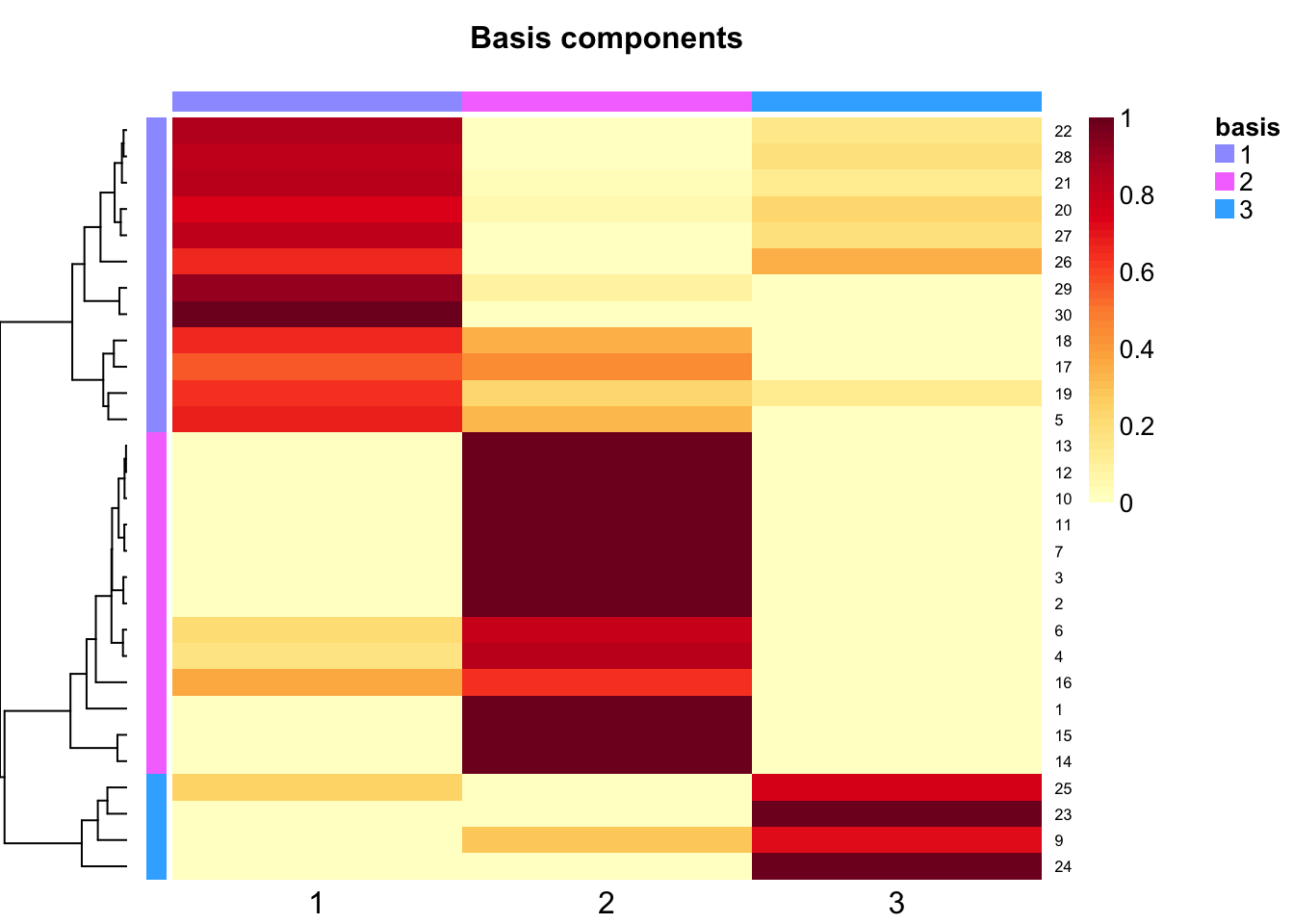
サイト番号が小さいほど上流ですので、basis 1が下流、basis2が上流に相当しています。basis 3は、25, 25, 9, 24というサイトからなっていますが、元の本によると、サイト23〜25は汚染があったところということで、それを反映したように見えます。サイト9は、本の図4.19をみると、サイト24にも近いようにみえるので、NMFではそちらに入ったのかもしれません。
次に、各魚種の潜在パターンへの分解(P)をヒートマップでしめします。
coefmap(spe_nmf)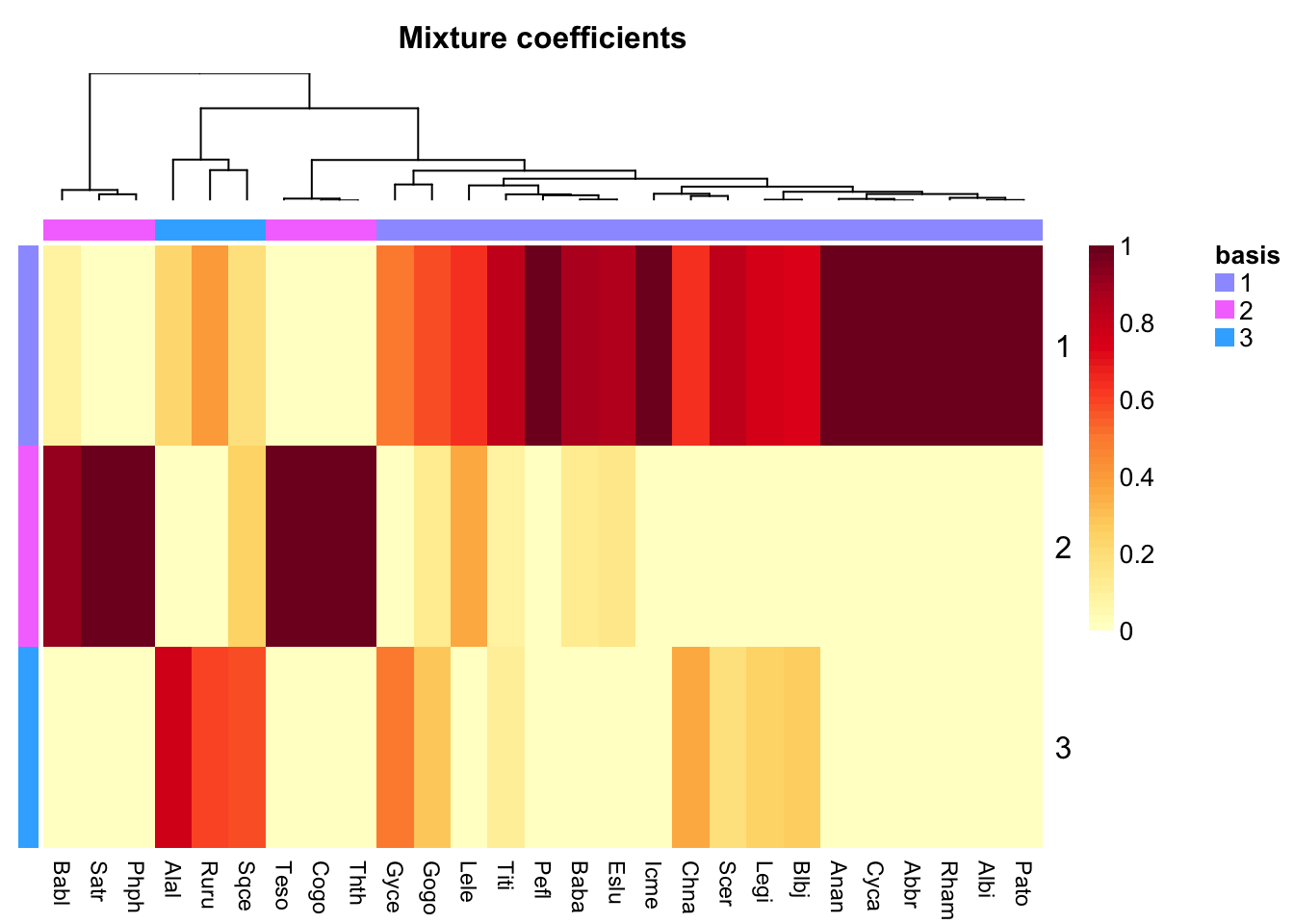
BablやSatr, Phphなど6種がbasis 2(上流)に多いというのは、本の図4.19でも同様です。一方basis 1(下流)の方が魚種がおおいというのも本の図4.19と同様です。
結論としては、わりとうまく分類ができたようです。